Oman_NGS
Mycobacterium tuberculosis example dataset
Table of contents
1. Introduction
The goal of this exercise is look map short reads (Illumina) to a reference genome and call variants. Our dataset are two M. tuberculosis samples that have been sequenced via Illumina paired-end. We will be using the trimmed reads from the previous module as input for the mapping.
We will
- Create a new
condaenvironment forsnippy - Use
snippyto map and call variants for each dataset - Look at SNPs
Map and call variants using snippy
First move into the TB dataset folder:
cd TB_module
We need to install snippy by creating a new conda environments
Install mamba by using the following command:
conda install -y -c conda-forge mamba
-y : automatically proceeds with downloading packages
-c conda-forge: adds conda-forge channel
mamba at the end are the names of the packages we are downloading mamba
If you previously installed the snippy environment please remove it here first:
conda remove --name snippy --all
Now we are going to use mamba to install snippy
mamba create --name snippy -c conda-forge -c bioconda snippy==4.6 snpeff==5.0 bcftools==1.10 vt==0.57721
mamba is a faster alternative to conda that sometimes works a bit better for some installations. It works in exactly the same way as conda.
mamba create : command that creates a new environment
-c conda-forge: adds conda-forge channel
-c bioconda: adds bioconda channel
snippy==4.6 snpeff=5.0 bcftools=1.10 vt==0.57721 : installs specific packages and versions
Once it has finished installing you can now activate the environment by typing:
conda activate snippy
This should do the trick, and everything should work now.
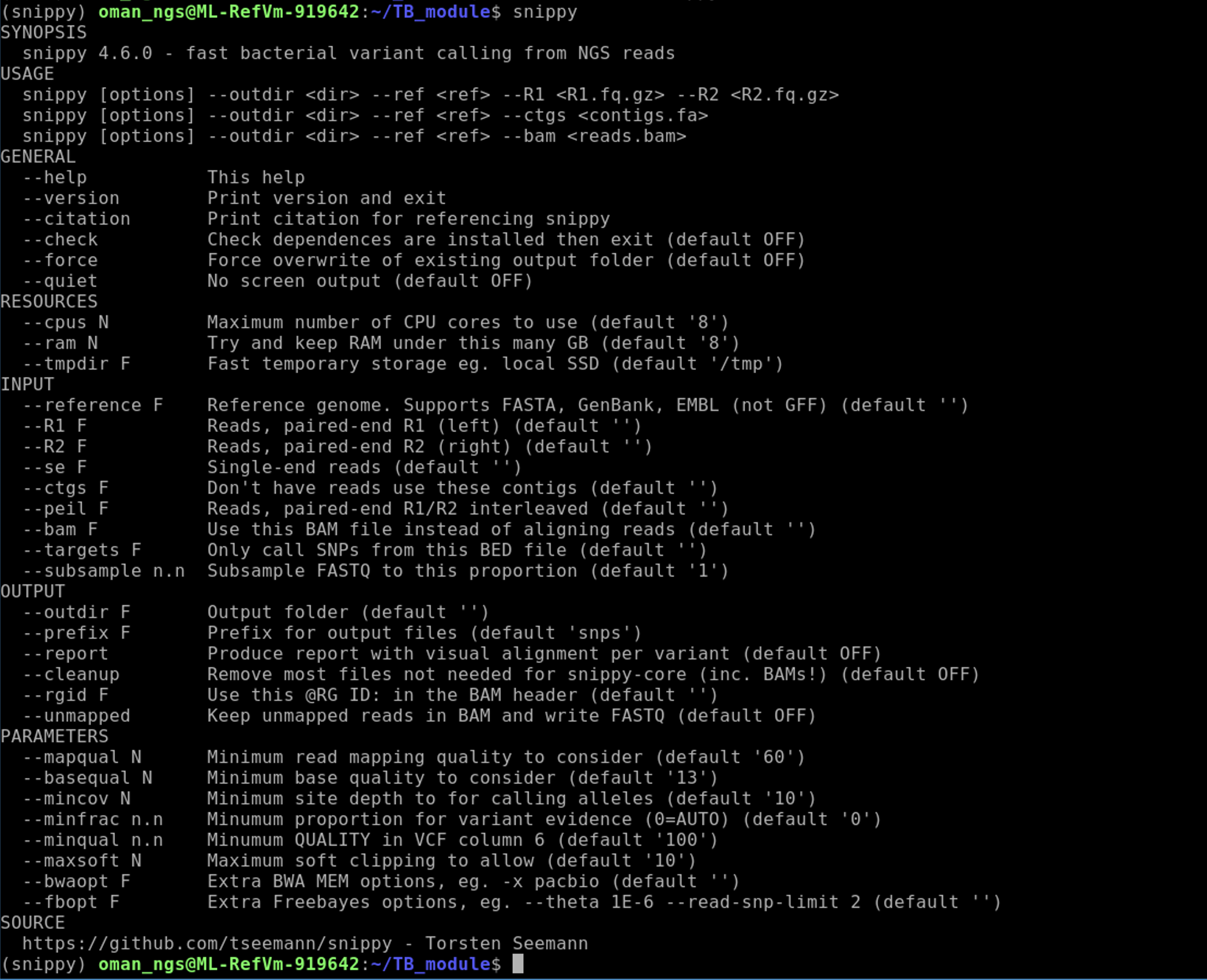
Check snippy is installed properly and to get an idea of how to run the program:
snippy -h
2. Run snippy to map reads to reference and call variants
We are going to use snippy which is a very popular pipeline for mapping and calling variants for bacterial genomes.
You can learn more about the pipeline here : https://github.com/tseemann/snippy
Let’s run snippy on the first TB sample:
snippy --cpus 4 --outdir TBsample1_snippy --ref Mtb_H37Rv.gb --R1 TBsample1_1_val_1.fq.gz --R2 TBsample1_2_val_2.fq.gz --subsample 0.5
--cpus 4 : run using 4 CPUs (the max number we have on the VMs). Default is 8.
--outdir : name the output directory
--ref : reference genome file in GenBank or fasta format
--R1 : Forward read files (remember we are using the trim_galore output files)
--R2 : Reverse read files (remember we are using the trim_galore output files)
--subsample : percentage to subsample reads. In this case we look at 0.5 (half) to speed things up. (You don’t have to do this with your actual data).
snippy will take a few minutes to complete. There will be quite a bit of output on the terminal – and that is OKAY!
When finished if you run:
ll TBsample1_snippy
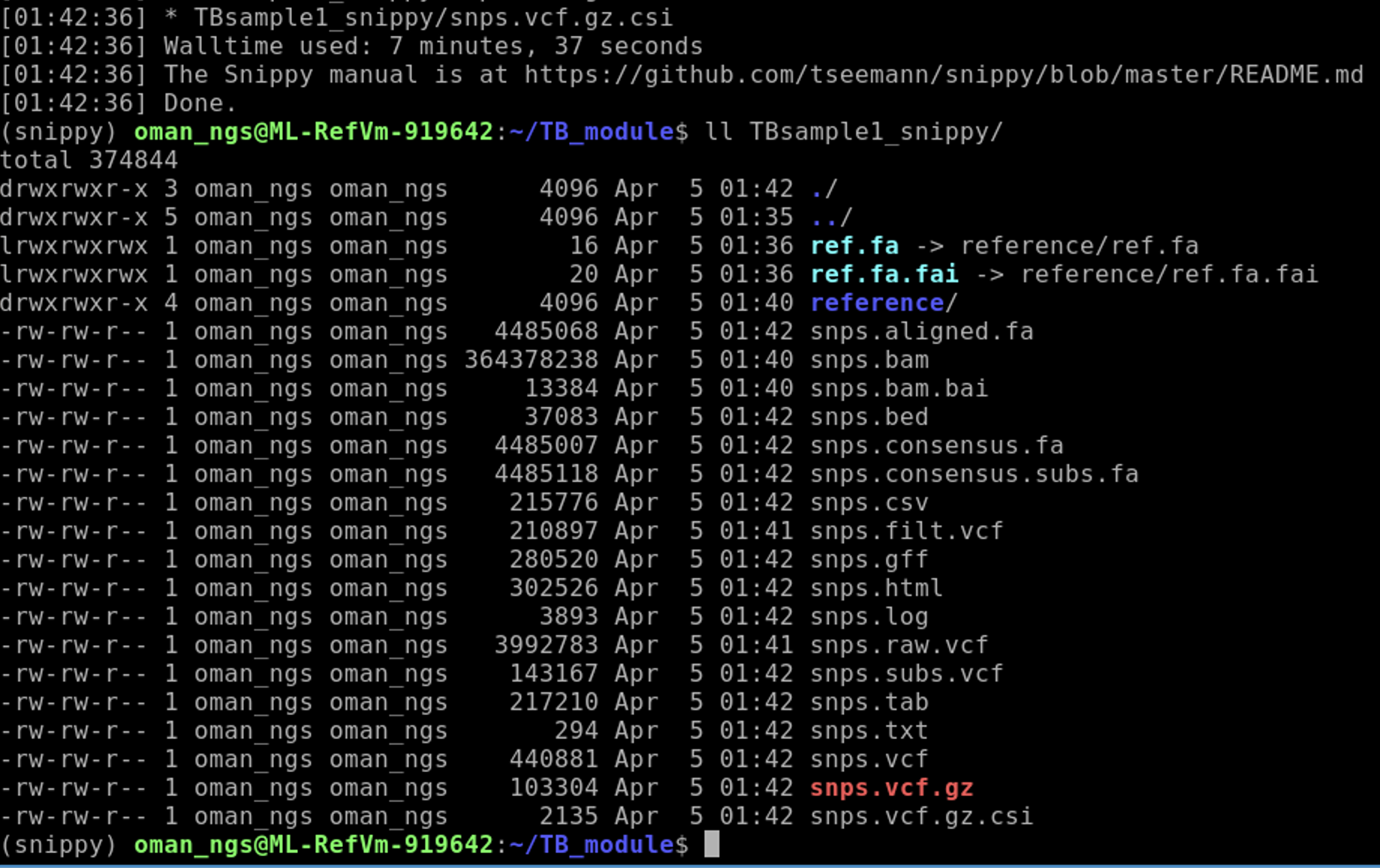
Now modify the command to run date for TBsample2.
How do you need to change the above snippy command so that you use that data?
Set --outdir TBsample2_snippy. What read data (--R1 and --R2) do we need to add?
After you have run both snippy commands you should see the following files is you run:
ls *snippy
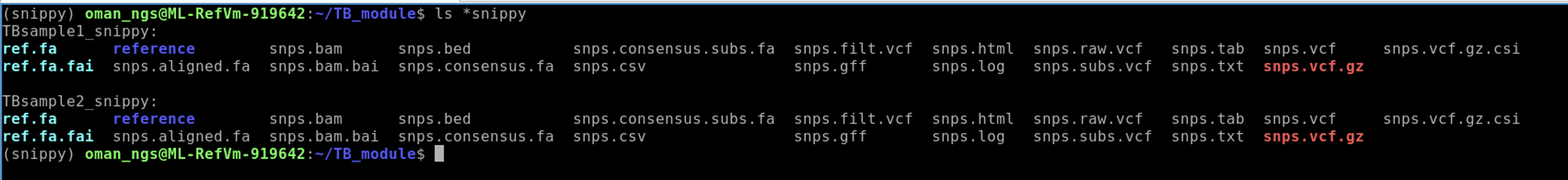 You should see both datasets that have the same files.
You should see both datasets that have the same files.
Looking at SNPs
There are many different files produced by snippy.
Output Files
| Extension | Description |
|---|---|
| .tab | A simple tab-separated summary of all the variants |
| .csv | A comma-separated version of the .tab file |
| .html | A HTML version of the .tab file |
| .vcf | The final annotated variants in VCF format |
| .bed | The variants in BED format |
| .gff | The variants in GFF3 format |
| .bam | The alignments in BAM format. Includes unmapped, multimapping reads. Excludes duplicates. |
| .bam.bai | Index for the .bam file |
| .log | A log file with the commands run and their outputs |
| .aligned.fa | A version of the reference but with - at position with depth=0 and N for 0 < depth < --mincov (does not have variants) |
| .consensus.fa | A version of the reference genome with all variants instantiated |
| .consensus.subs.fa | A version of the reference genome with only substitution variants instantiated |
| .raw.vcf | The unfiltered variant calls from Freebayes |
| .filt.vcf | The filtered variant calls from Freebayes |
| .vcf.gz | Compressed .vcf file via BGZIP |
| .vcf.gz.csi | Index for the .vcf.gz via bcftools index) |
Let’s take a look at a summary file to see how many variants were called in total:
cat TBSample1_snippy/snps.txt
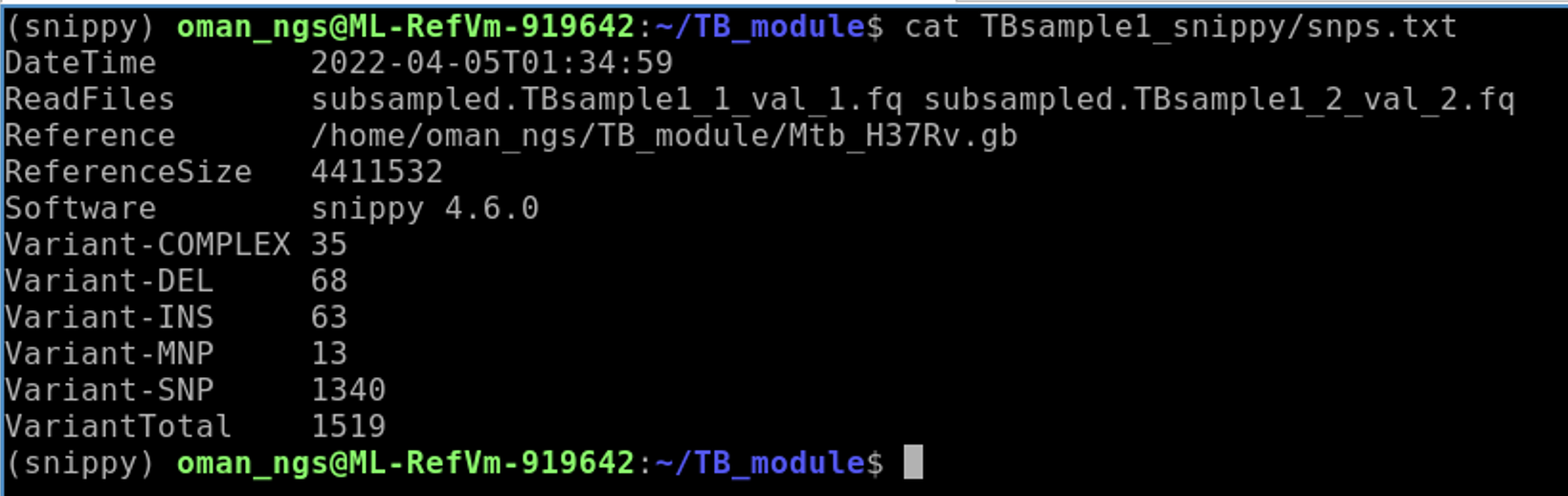 How many variants total were called against for this sample?
How many variants total were called against for this sample?
Look at the snps.txt for TBSample2.
cat TBSample2_snippy/snps.txt
Which TB dataset has more variants called?
Looking at the snps.html file
One of the most useful files for us to look at is the snps.html file. This file will list each of the mutations found and describe what gene and what type of mutation – for instance whether it is a synonymous or non-synonymous mutation. If a non-synonymous mutation it will list the amino acid difference and the gene effected.
Open the following file:
firefox TBsample1_snippy/snps.html
Columns in the TAB/CSV/HTML file formats
Name | Description
—–|————
CHROM | The sequence the variant was found in eg. the name after the > in the FASTA reference
POS | Position in the sequence, counting from 1
TYPE | The variant type: snp msp ins del complex
REF | The nucleotide(s) in the reference
ALT | The alternate nucleotide(s) supported by the reads
EVIDENCE | Frequency counts for REF and ALT
If you supply a Genbank file as the --reference rather than a FASTA
file, Snippy will fill in these extra columns by using the genome annotation
to tell you which feature was affected by the variant:
| Name | Description |
|---|---|
| FTYPE | Class of feature affected: CDS tRNA rRNA … |
| STRAND | Strand the feature was on: + - . |
| NT_POS | Nucleotide position of the variant withinthe feature / Length in nt |
| AA_POS | Residue position / Length in aa (only if FTYPE is CDS) |
| LOCUS_TAG | The /locus_tag of the feature (if it existed) |
| GENE | The /gene tag of the feature (if it existed) |
| PRODUCT | The /product tag of the feature (if it existed) |
| EFFECT | The snpEff annotated consequence of this variant (ANN tag in .vcf) |
Variant Types
| Type | Name | Example |
|---|---|---|
| snp | Single Nucleotide Polymorphism | A => T |
| mnp | Multiple Nuclotide Polymorphism | GC => AT |
| ins | Insertion | ATT => AGTT |
| del | Deletion | ACGG => ACG |
| complex | Combination of snp/mnp | ATTC => GTTA |
3. Looking for resistance mutations
Let’s look for a few well known resistance mutations in these datasets.
Rifampin reseistance
A primary drug for treating TB is rifampin. One of the main genes that has mutations that confers resistance to rifampin is rpoB. Let’s take a look to see if we have any mutations in rpoB within our two samples.
grep 'rpoB' TBsample1_snippy/snps.tab
grep 'rpoB' TBsample2_snippy/snps.tab
grep is the command to search within a file
Here, rpoB after grep is the search terms and then we follow it by the file we wish to search.
Does one sample have more mutations than the other?
One particular mutation in rpoB that is known to confer resistance to rifampin is the S640L mutation. Does one of our samples have this mutation?
Isoniazid resistance
Take a look at this paper here: https://academic.oup.com/jid/article/182/6/1788/916586?login=false
What gene and what mutation within that gene are typical for isoniazid resistance with TB?
Use grep to search for this gene within the TBsample1_snippy/snps.tab and TBsample2_snippy/snps.tab