Oman_NGS
Table of contents
1. Introduction
In this module, we are going to work through how to generate SARS-CoV-2 consensus genomes from Oxford Nanopore data. The most popular pipeline for this is called ARTIC.
You can find more information here:
ARTIC : https://github.com/artic-network/fieldbioinformatics
This data was generated by sequencing on an ONT GridION using Midnight V1200 primer scheme. Because ONT can produce much longer reads, the amplicon size can greatly be increased on these devices.
See more in comparison with the V4 scheme here : https://nanoporetech.com/covid-19
First we need to download the SARS-CoV-2 files:
cd Oman_modules
git pull
Check to see we now have the SARS_CoV-2 folder:
ll
1. Install the ARTIC Bioinformatics
A very popular pipeline for processing SARS-CoV-2 nanopore sequencing data is ARTIC. They put out an SOP for running the pipeline back in 2020, and it is still quite useful to follow:
https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html
It is always a good idea to keep mamba up to date, so we will first run:
mamba update mamba
We will follow the directions to install ARTIC from here:
cd SARS_CoV-2
cd artic-ncov2019
## This step will take a few minutes:
mamba env create -f environment.yml
conda activate artic-ncov2019
2. Using tar to uncompress folders
Now move back into the SARS_CoV-2 folder to use tar to uncompress the files:
cd ../SARS_CoV-2
ll
You will see that there are three barcode files that end in .tar.gz. This means these are folders that are gzipped (.gz) or compressed. We need to uncompress them for us to use them.
Use tar to uncompress:
tar xzvf barcode01.tar.gz
ll
tar [options] [compressed-file] [file or directory to be compressed] :
-x : Extract (decompress)
-c : compress
-z : zip, tells tar command to create or uncompress tar file using gzip
-v : Displays Verbose Information (print what is happening to the screen)
-f : creates archive with given filename
tar xzvf [compressed file] is standard way to uncompress
To compress a folder use similar command:
tar czvf [<file_name>.tar.gz] [file or folder]
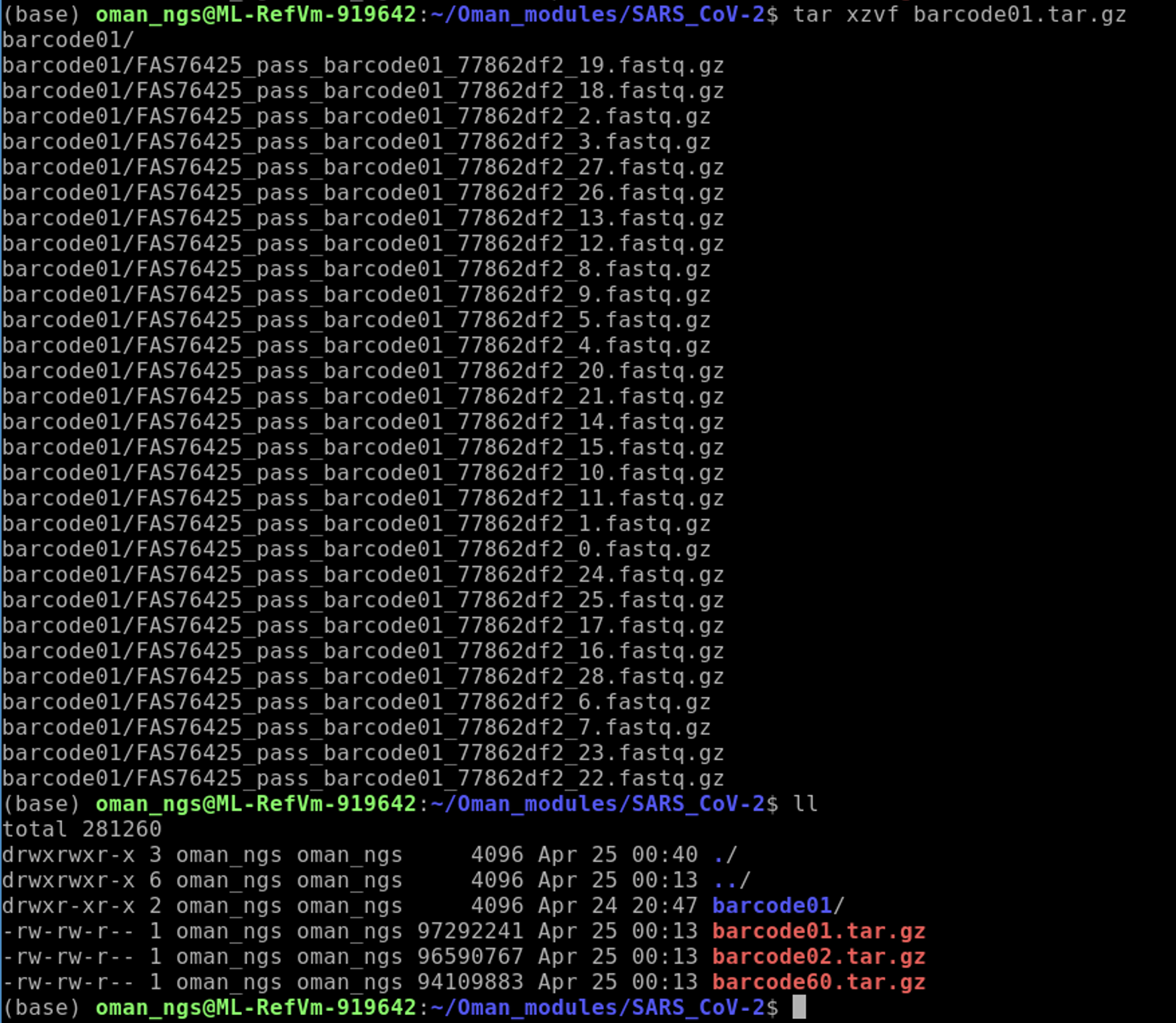
You should see the barcode01 folder now.
Use the tar command to uncompress the other two barcode file
After, you will have the following three barcode folders:
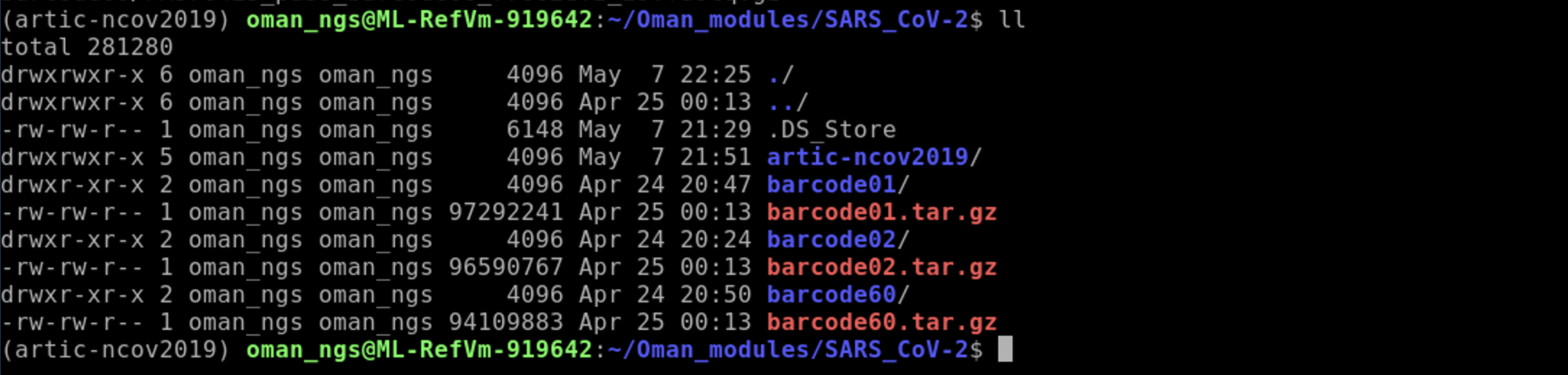
After you have unzipped all of the folders, you are free to delete the .gz files. We are running tight on space on the VMs so this will help!
rm *.gz
Note if you run out of disk space !!!!
If you run out of disk space on the VM you can delete the TB raw fastq files as these are very large files.
cd ~/TB_module
rm *.fastq.gz
3. ARTIC bioinformatics workflow:
We are picking up the protocol at the guppyplex command, after the base-calling and demultiplexing steps. Instructions for those are in the SOP :
https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html
First we make a directory for the output files of the guppyplex command:
mkdir guppyplex
Now since we are using the Midnight primer set, which has amplicons up to 1200bp, we will run the guppyplex command like so:
artic guppyplex --skip-quality-check --min-length 250 --max-length 1500 --directory barcode01 --prefix guppyplex/run01
--skip-quality-check : we are using only the ‘passed’ reads from the run (high quality) and therefore as per the protocol we can skip this step and speed up the pipeline a bit. You do not have to use this option if you don’t want in your real analyses.
--min-lenght <value_in_bp> : the minimum read length used (for traditional ARTIC V4 this is set at 400)
--max-lentgth <value_in_bp>: the maximum read length used (for traditional ARTIC V4 this is set at 700)
--directory : dir name containing the fastq files to process
--prefix : a prefix (usually the run name) that is added at the beginning of all fastq files as output. Here we specify the output folder guppyplex and then run01 as a prefix

Please run this command for the other two samples (barcodes)
Run artic minion pipeline which is the alignment/variant-call/consensus pipeline
We are running the medaka pipeline, which only uses the fastq files. The nanopolish version of this pipeline uses the raw fast5 output files for variant calling. These fast5 files are very large, which is why we can’t use them in our VMs. In general, these pipelines will produce identical results for SARS-CoV-2 data.
artic minion <options> <primer_scheme> <sample>
artic minion --strict --threads 4 --medaka --medaka-model r941_min_high_g360 --scheme-directory artic-ncov2019/primer_schemes --read-file guppyplex/run01_barcode01.fastq nCoV-2019/V1200 run01_barcode01
| Argument name(s) | Required | Default value | Description |
|---|---|---|---|
| scheme | Y | NA | The name of the primer scheme |
| sample | Y | NA | The name of the sample |
| –medaka | N | False | Use medaka instead of nanopolish for variants |
| –medaka-model | * | NA | Medaka model to use (required if –medaka set) |
| –minimap2 | N | True | Use minimap2 |
| –bwa | N | False | Use bwa instead of minimap2 |
| –normalise | N | 100 | Normalise down to moderate coverage to save runtime |
| –threads | N | 8 | Number of threads |
| –scheme-directory | N | /artic/schemes | Default scheme directory |
| –max-haplotypes | N | 1000000 | Max-haplotypes value for nanopolish |
| –read-file | N | NA | Use alternative FASTA/FASTQ file to |
| –fast5-directory | N | NA | FAST5 Directory |
| –sequencing-summary | N | NA | Path to Guppy sequencing summary |
| –skip-nanopolish | N | False | Skip nanopolish |
| –no-longshot | N | False | Use medaka variant instead of longshot (experimental feautre from v1.2.0) |
| –strict | N | False | Enables experimental features (from v1.2.0), including VFC overlap checks and stats |
| –dry-run | N | False | Perform a dry run of the minion pipeline, outputing commands to a log but not executing them |
More on medaka-models: https://github.com/nanoporetech/medaka#models
Medaka models are named to indicate i) the pore type, ii) the sequencing device (MinION or PromethION), iii) the basecaller variant, and iv) the basecaller version, with the format:
{pore}_{device}_{caller variant}_{caller version}
r941_min_high_g360
In our case we sequenced on Minion/GridION R9.4.1 flowcell using the “high” accuracy base-calling mode and the latest version of guppy that this supports was version 3.6.0.
! Please amend the command above and run the other two samples through the artic minion pipeline
There are many output files produced by the pipeline. I will list the most important ones here, but you can find the full description of all files here : https://artic.readthedocs.io/en/latest/minion/
Output files:
Mapping files:
| file name | description |
|---|---|
$SAMPLE.sorted.bam |
the raw alignment of sample reads to reference genome |
$SAMPLE.trimmed.rg.sorted.bam |
the post-processed alignment |
$SAMPLE.primertrimmed.rg.sorted.bam |
the post-processed alignment with additional softmasking to exclude primer sequences. Use this file for reading into Artemis / IGV |
Called variants files:
| file name | description |
|---|---|
$SAMPLE.$READGROUP.vcf |
the raw variants detected (one file per primer pool) |
$SAMPLE.merged.vcf |
the raw variants detected merged into one file |
$SAMPLE.vcfreport.txt |
a report evaluating reported variants against the primer scheme |
$SAMPLE.fail.vcf |
variants deemed too low quality |
$SAMPLE.pass.vcf.gz |
detected variants (indexed) |
Consensus genome files:
| file name | description |
|---|---|
$SAMPLE.*_mqc.json |
stats files which MultiQC can use to make a report |
$SAMPLE.consensus.fasta |
the consensus sequence for the input sample |
$SAMPLE.muscle.out.fasta |
an alignment of the consensus sequence against the reference sequence |
4. Identify primer dropouts using MultiQC
Once you have run each sample through the artic minion pipeline, please run the following:
Remember the dot at the end of this command
multiqc .
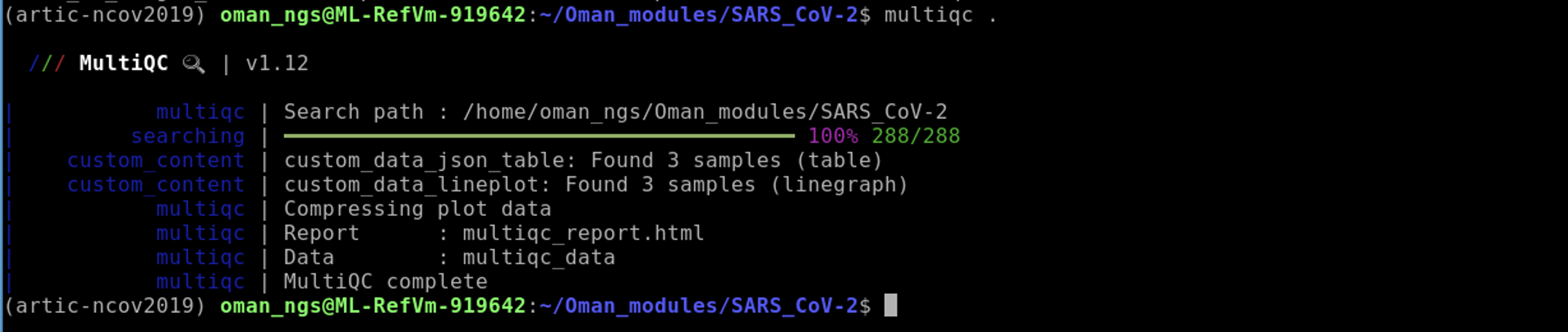
Then run:
firefox multiqc_report.html
Questions
- Are there any amplicon drop outs in the samples?
- Which amplicon is it?
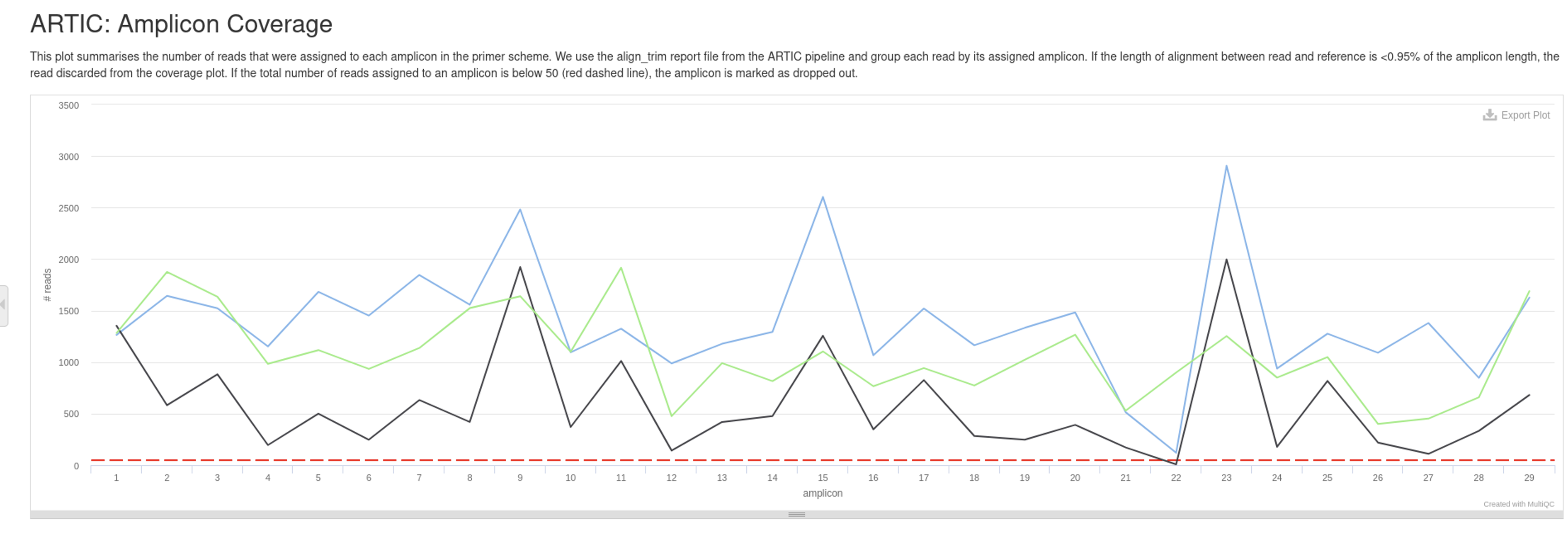
5. Use IGV to view mapped reads (bam file) to see why we have a dropout
We have already used Artemis as a genome viewer, but another very popular one is the Integrated Genome Viewer, or IGV, from the Broad Institute.
Website here : https://igv.org/
I had previously installed igv within the base conda environment. Therefore you can start IGV like so:
conda activate base
igv
First we need to download the SARS-CoV-2 reference
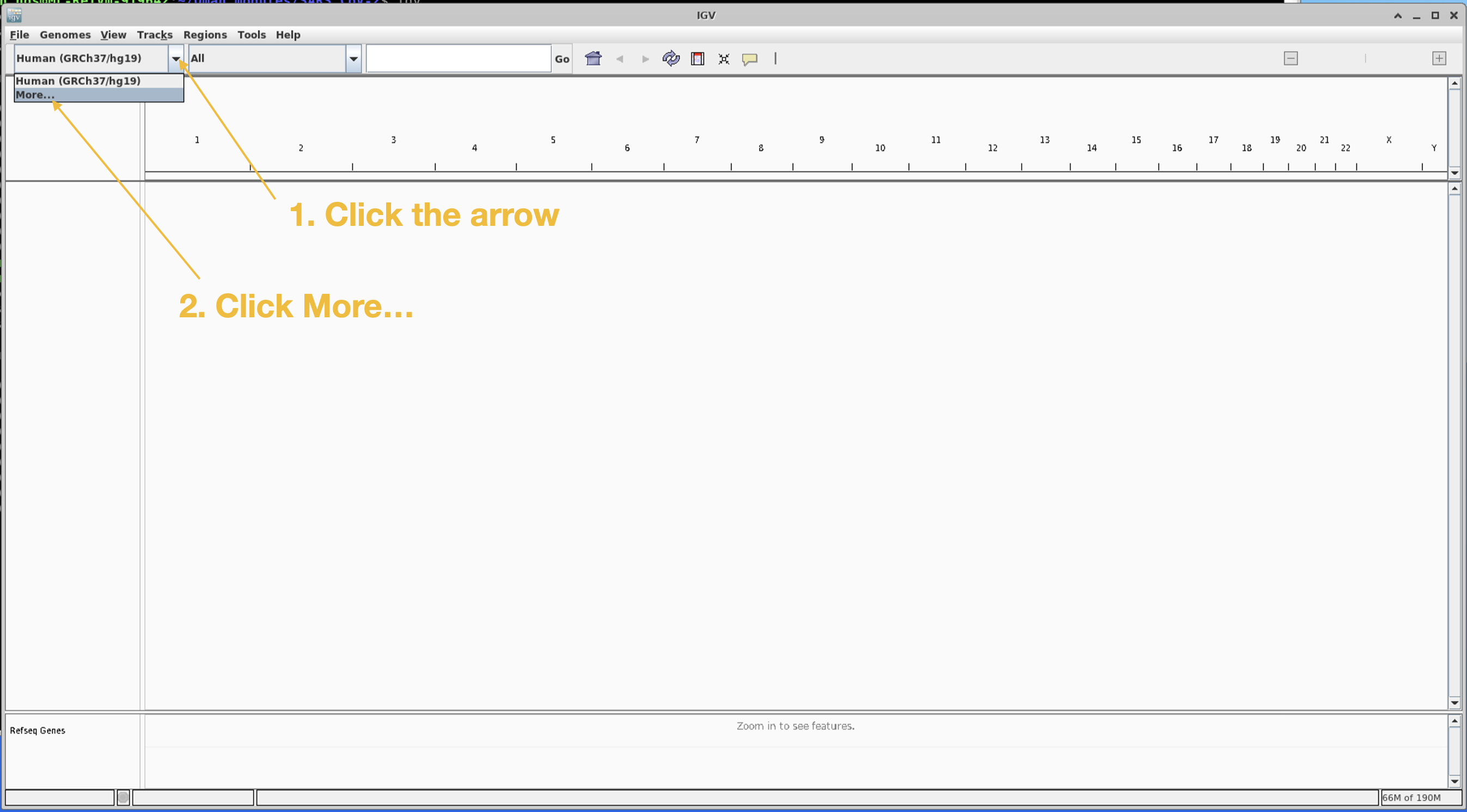
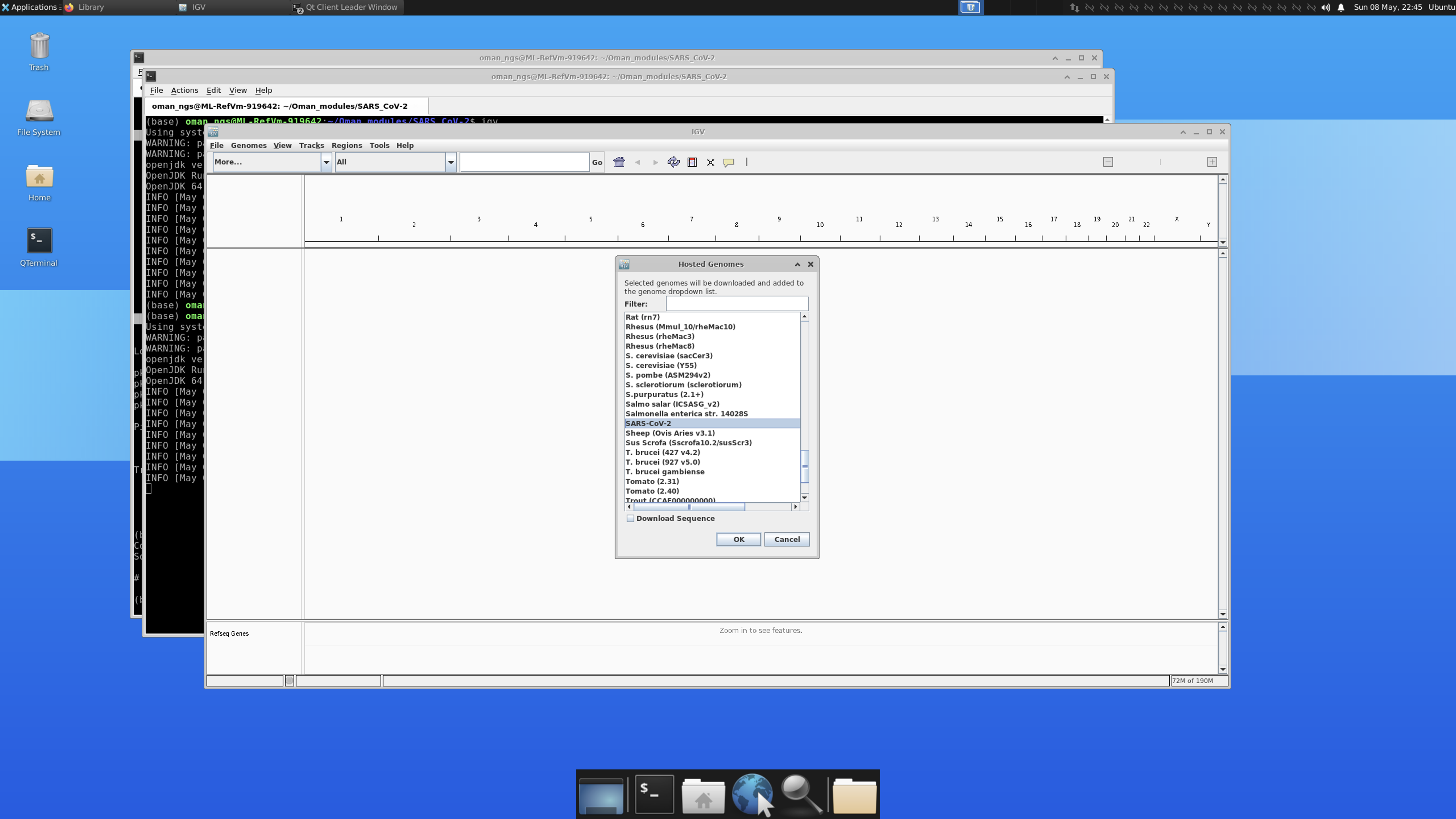
Scroll down the pop-up list until you find SARS-CoV-2. Click SARS-CoV-2 and click OK.
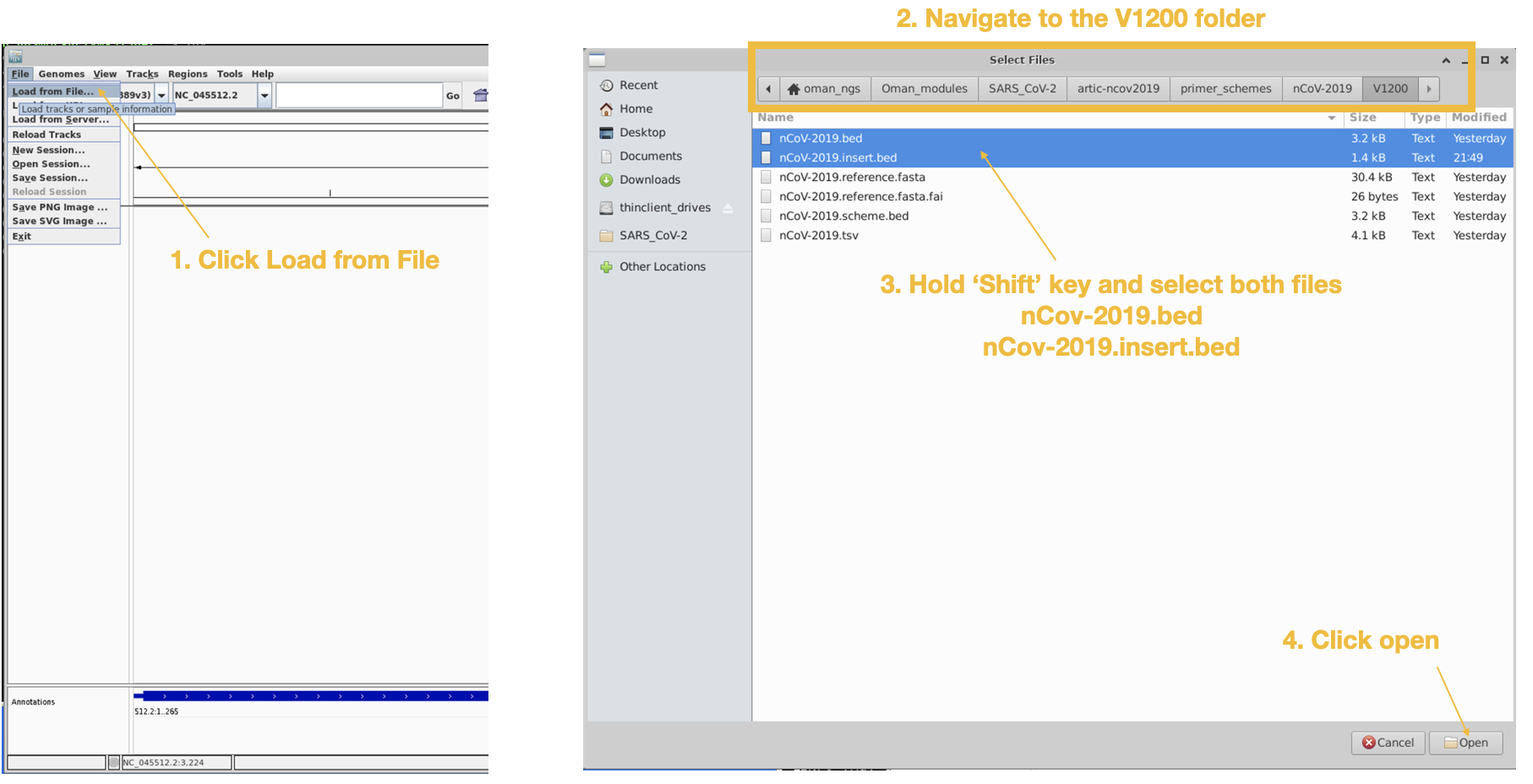
Next we will load in the amplicon are the the primer binding site files.
We can view the amplicons easier by changing the view this way:
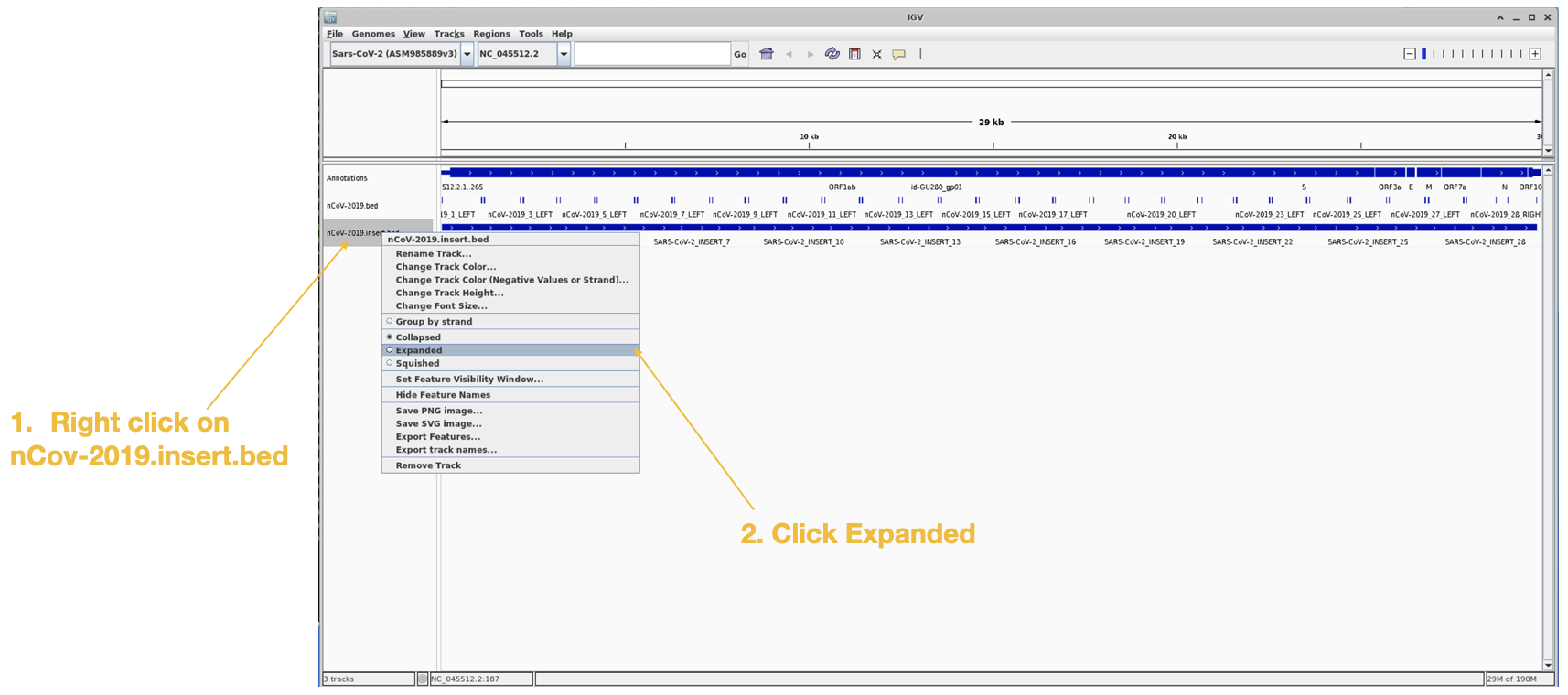
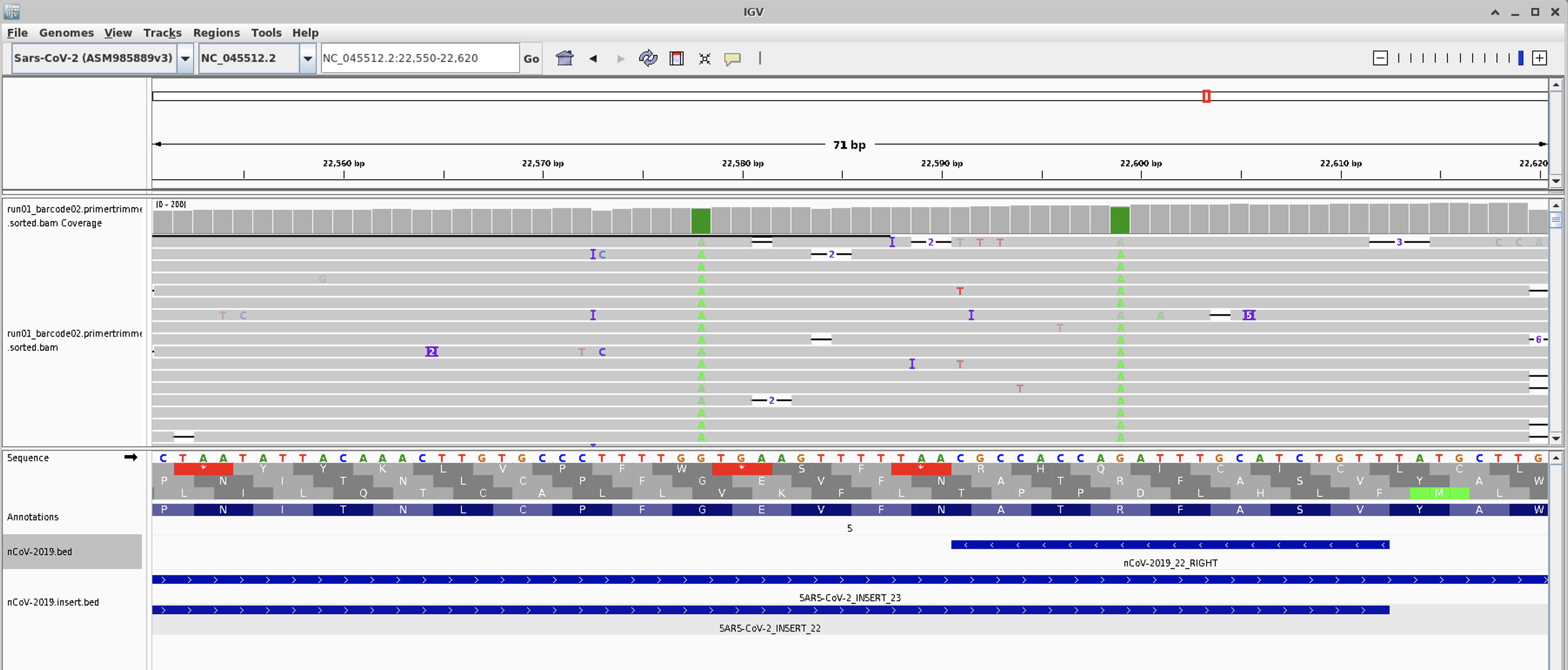
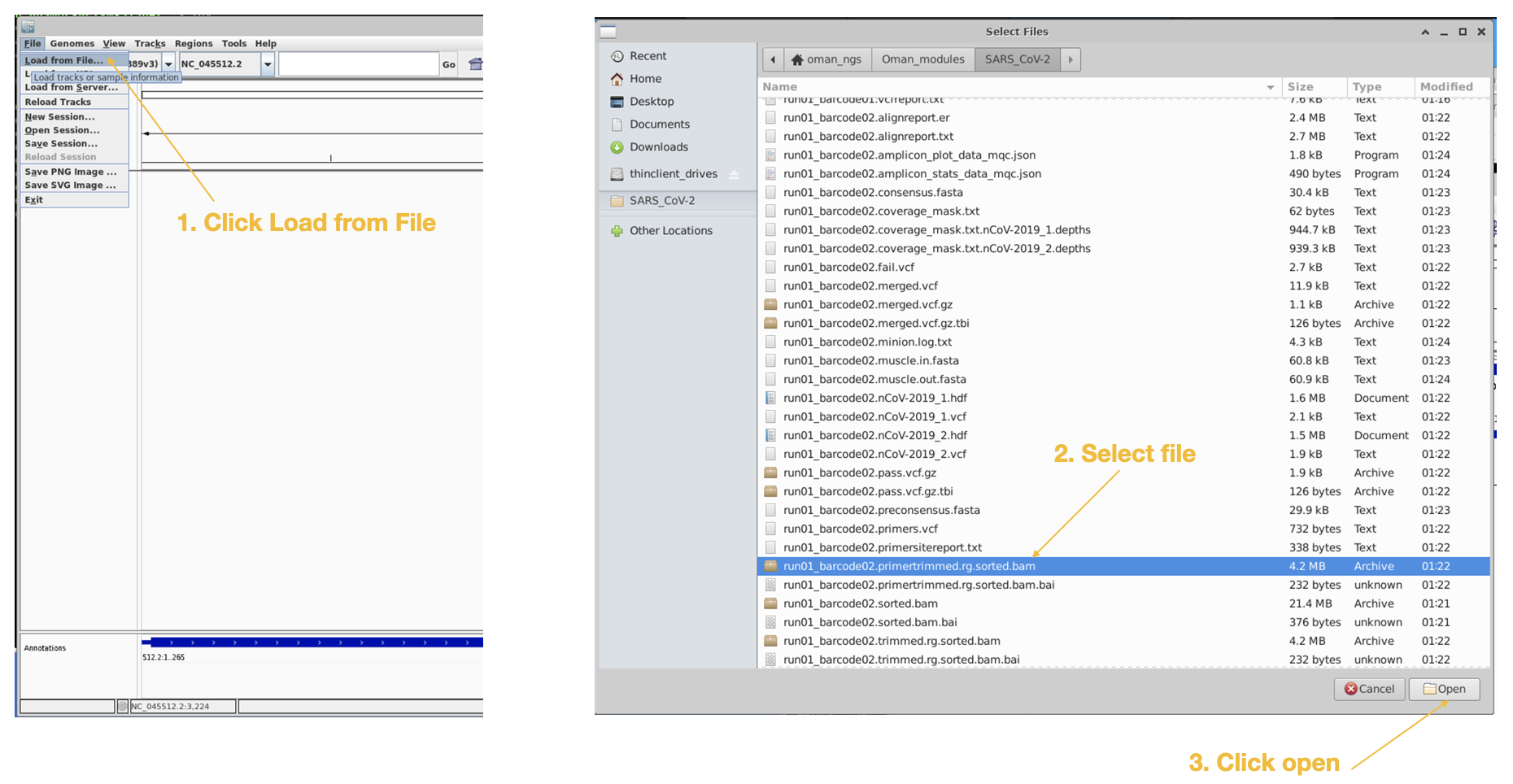
Lastly we will load in the primertrimmed.rg.sorted.bam file for barcode02, since that one showed the dropout.
You should now see all the reads being displayed:
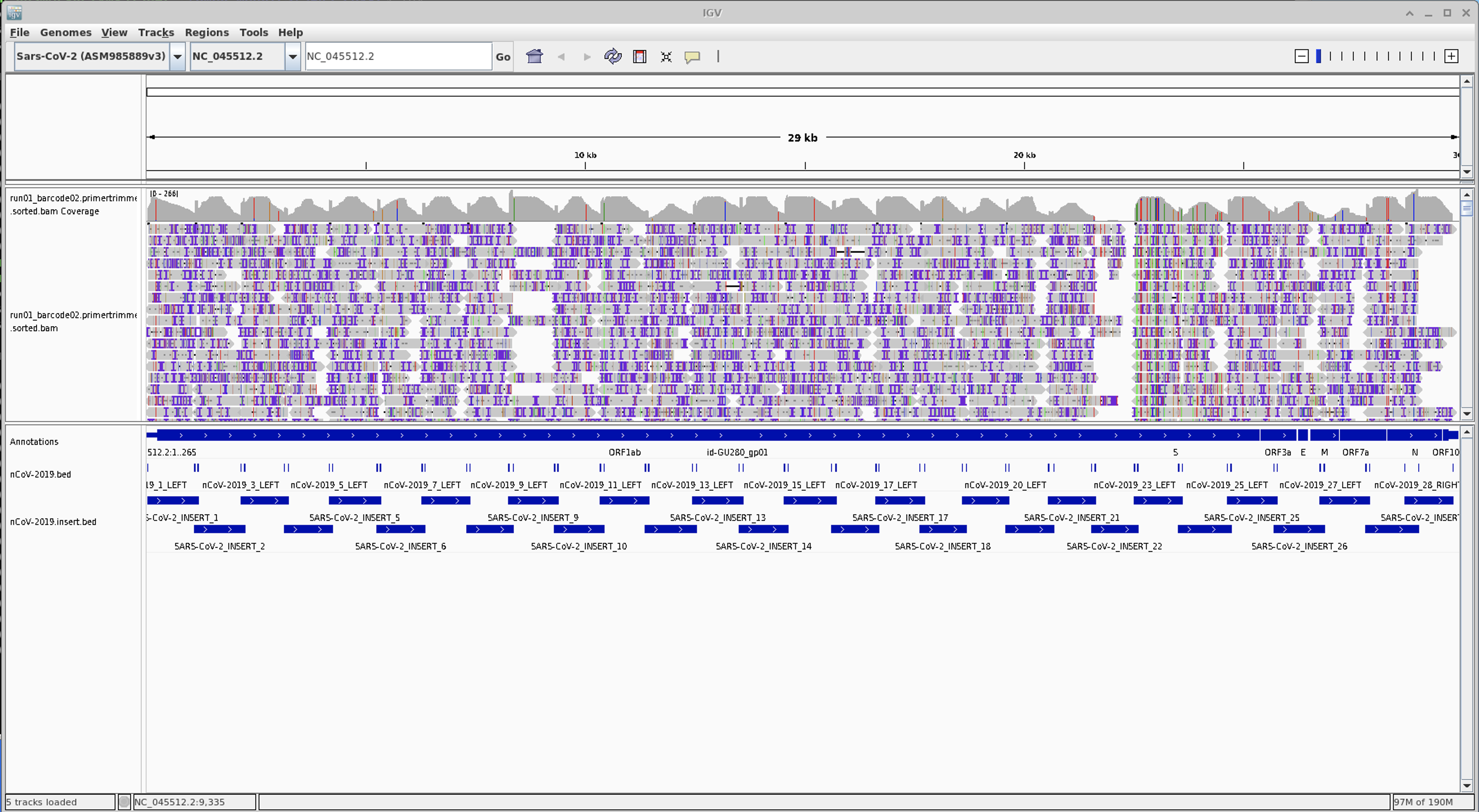
Zoom in on amplicon 22
Questions
- Do you see any mutations occuring in amplicron 22 primer sites?